





























































































 免費參與·100+跨境活動
免費參與·100+跨境活動 
 免費下載·4000+跨境資料
免費下載·4000+跨境資料 
 免費學習·2000+直播課程
免費學習·2000+直播課程 
 免費加入·15萬+賣家交流群
免費加入·15萬+賣家交流群 


2023-07-05 16:50
 圖片來源:跨境白武士 Jaems
圖片來源:跨境白武士 Jaems
全球速賣通發布關于加強出口歐盟商品資質管控公告,公告如下:
 圖片來源:全球速賣通
圖片來源:全球速賣通
商品合規一直是速賣通關注重點,為了應對當下全球消費者的需求增長,保障商家安全公平的交易環境、提升消費者體驗以及滿足各國合規和安全要求,速賣通要求商家發布的商品符合目的市場的相關法律及規定。任何銷往歐洲地區的商品需要嚴格符合歐盟相關的商品合規及商品安全要求。
平臺擬進一步加強售往歐盟的嬰幼兒玩具、電子電器、小家電、裝飾燈具、醫療器械、化妝品、珠寶飾品、運動戶外、安全防護等品類的商品管控。商家需確保自己提供的商品符合銷售國當地的相關法律法規。
具體類目的合規要求,平臺將結合各國合規及法律安全要求,對于不滿足目的國合規要求的商品(包括但不限于缺失CE證書,EMC/LVD檢測報告,ROHS檢測報告及商品外包裝標簽圖(包含產品信息、生產企業信息、歐代責任人信息以及資質Logo及必要警示語)等,速賣通可能對相關商品屏蔽區域化市場,對于惡意規避的商品將執行下架扣分等處罰。
今天讓我們來了解下醫療器械出口法規須知。
法規背景:
醫療器械在美國上市主要需根據美國聯邦法規法典 第21冊 - 食品和藥品中的要求進行管控,該法規規定了醫療器械的定義,分類,管控方式。其他還可能涉及到的法規有:
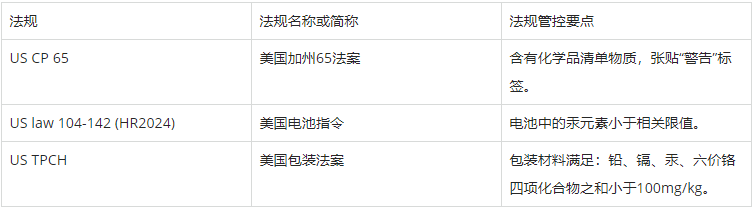
US law 104-142 (HR2024)指令規定了美國境內電池產品相關管控要求。指令規定了相關電池產品相關有害物質限制要求。
US TPCH法案規定了美國境內包裝材料的相關管控要求。法案規定了包裝材料相關有害物質的限制要求。
醫療器械在歐盟上市主要需符合以下法規:
預期取代醫療器械指令Medical Devices Directive COUNCIL DIRECTIVE 93/42/EEC和有源植入醫療器械指令AIMD Council Directive 90/385/EEC
預期取代體外診斷醫療器械指令 IVDD,Council Directive 98/79/EEC
法規規定了醫療器械的定義,分類規則,及合規流程。
其他還可能涉及到的法規有:
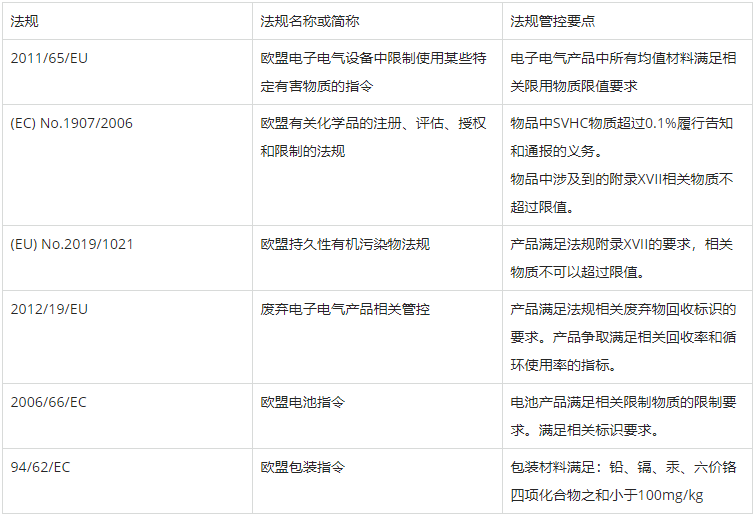
(EC) No.1907/2006法規規定了歐盟境內生產、制造和銷售所有物質、混合物和物品的相關化學品限制要求。相關物品需要滿足法規Article 33關于SVHC的要求以及Article 67關于附錄XVII的要求。
(EU) No.2019/1021法規規定了歐盟境內所有物質、混合物和物品中相關持久性有機污染物的限制要求。
2012/19/EU指令規定了歐盟境內廢棄電子電氣產品相關管控要求。指令涉及到回收標識和回收率等相關規定。
2006/66/EC指令規定了歐盟境內相關電池產品的相關管控要求。指令涉及到相關限用物質限制要求和標識要求。
94/62/EC指令規定了歐盟境內相關包裝材料的相關管控要求。指令規定了包裝材料相關有害物質的限制要求。
管控范圍:
美國的醫療器械是指符合以下條件的儀器、裝置、工具、機具、器具、插入管、體外試劑或其它相關物品,包括組件、零件或附件等;意圖使用于人類疾病或其它身體狀況的診斷;或用于疾病之治愈、減緩、治療者;意圖影響人類身體的功能或結構,但不經由人類身體或身體上的化學反應來達成其首要目的,同時也不依賴新陳代謝來達成其主要目的。
歐盟的醫療器械新法規對醫療器械的定義修改如下:是指單獨或者組合使用于人體的任何儀器、設備、器具、軟件、植入物、試劑、材料或其他物品;其作用于人體體表或體內的主要效用不是通過藥理學、免疫學或者代謝的手段獲得,但可能有這些手段參與并起一定的輔助作用;旨在達到下列一個或多個目的:疾病的診斷、預防、監護、治療或緩解;損傷或者殘疾的診斷、監護、治療、緩解或修補;解剖學和生理或病理過程或狀態的探查、替代或調節;來自器官、血液和組織捐獻的人體標本體外檢驗數據的提供;專門用于對醫療器械清洗、消毒或滅菌和對妊娠的控制和支持的器械應被認為是醫療器械。
法規要求:
1. 美國:
美國聯邦法規法典第 21 冊-食品和藥品中對醫療器械按照風險等級做了分類,低風險的器械歸為I類,中等風險的器械歸為II類,高風險的器械歸為III類。針對具體器械進行了規則編號,且每一種器械指定了器械代碼,并指定了產品需要符合的相關標準。
a. 低風險I類
I類為“普通管理”產品,是指危險性小或基本無危險性的產品,它的設計一般比Ⅱ類產品、Ⅲ類產品簡單。FDA 認為絕大多數的 I 類產品通過一般控制足以保證其安全性和有效性。I 類產品要求符合一般控制,例如醫用手套、壓舌扳、手動手術器械等。管控要求為一般管控,即在FDA進行注冊列名。
b. 中風險Ⅱ類
Ⅱ類是指那些用一般控制不足以控制其安全性和有效性,必須通過現有的其他方式,即特殊控制,來保證其安全性和有效性的產品。例如體溫計,心電圖儀、超聲診斷儀、輸血輸液器具、呼吸器等。在Ⅱ類產品市場準入前,一般需申請市場準入前通告 510(k)。
c. 高風險Ⅲ類
Ⅲ類是指那些僅用一般控制和特殊控制還不足以確保其安全性和有效性的產品。這類產品具有較大危險性或危害性,它一般用來支持人體生命,防止人體健康受損,具有致病、致殘的潛在的、不合理的風險。例如人工心臟瓣膜、心臟起搏器、人工晶體、人工血管等。FDA對這類產品實行上市前批準(PMA)制度。
另外,關于化學方面,還可能涉及:
 圖片來源:全球速賣通
圖片來源:全球速賣通
2. 歐盟:
歐盟醫療器械法規將醫療器械分成Ⅰ,Ⅱa,Ⅱb 和Ⅲ類等四個類別:
Ⅱa 類包括診斷設備、體液儲存、輸入器械,以及短暫使用(持續時間小于 1h)并有侵害性的外科器械。
Ⅱb 類為短期使用(持續時間 lh~30d)并有侵害性的外科用器械、避孕用具和放射性器械。
Ⅲ類器械為與中樞神經系統、心臟接觸的器械、在體內降解的器械、植入體內的器械和藥物釋放器械,以及長期使用(持續時間大于30d)并有侵害性的外科器械。
a. 低風險I類
醫療器械指令/法規明確規定了I類醫療器械的分類規則,需要符合的基本安全和性能要求及評估流程。I類醫療器械包括I類測量器械(Im),I類滅菌器械(Is),和I類其他器械;I類其他器械可以出具自我符合性聲明來滿足CE要求;I類測量器械(Im),I類滅菌器械(Is)則需要公告機構(Notified body)簽發CE認證證書。
b. 中風險II類
醫療器械指令/法規明確規定了II類醫療器械的分類規則,需要符合的基本安全和性能要求及管控要求。須向公告機構提出上市申請,由公告機構負責審查(Ⅱa 類產品的產品設計由生產企業負責,公告機構主要檢查其質量體系;Ⅱb 類產品由公告機構審查質量體系、抽檢樣品,同時生產企業應提交產品設計文件),通過審查后,簽發CE認證證書,產品貼 CE 標志,方可進入歐盟各成員國市場。
c. 高風險III類
醫療器械指令/法規明確規定了Ⅲ類醫療器械的分類規則,需要符合的基本安全和性能要求及管控要求。Ⅲ類產品由公告機構審查,要檢查質量體系、抽檢樣品,并審查產品設計文件,特別是審查產品風險分析報告。通過審查后,簽發CE認證證書,產品貼 CE 標志,方可進入歐盟各成員國市場。
另外,關于化學方面,還可能涉及:
 圖片來源:全球速賣通
圖片來源:全球速賣通
(來源:跨境白武士James)
以上內容屬作者個人觀點,不代表雨果跨境立場!本文經原作者授權轉載,轉載需經原作者授權同意。?
 收錄于以下專欄
收錄于以下專欄





















